




Project purpose

In this project, we try to create novel molecular databases based on computational chemistry methods, where molecules registered in PubChem are targeted. This project heavily uses quantum chemistry and first-principles methods, in addition to artificial intelligence techniques. We will also develop tools used together with our databases.
About 100 millions molecules are already registered in the PubChem database. The data is rich and diverse, and numerous chemically important molecules are treated. However, PubChem mainly provides the structure information of molecules, which are described by SMILES and InChI representations.
Various molecular properties need to be handled to widely use the database.
So, this project tries to expand its possibilities by using computational chemistry and information science techniques. Now, we are trying to add various molecular electronic structure properties, such as HOMO-LUMO gap and excitation energy, by using first-principles quantum chemistry methods.
We hope that our databases and tools can help to research various chemistry, such as materials, catalysts, drugs, and so on.
About us

Scientists specializing in computational (quantum) chemistry and information science are participating in this project.
Our project is driven by computational chemists Tomomi Shimazaki (Ten-no group@Kobe University) and
Maho Nakata (RIKEN),
and information scientists Toshiyuki Maeda (STAIR) and Masatomo Hashimoto (STAIR).
We will promote this project through collaborations of researchers in these two different areas.
Any questions for this site: pccdb (at) ml.riken.jp
PubChemQC project

PubChemQC provides the ground- and excited-state electronic structures, and targets molecules registered in PubChem.
At present, about 3.2 millions molecules, whose geometries (structures) are optimized by B3LYP/6-31G*, are provided. In addition, about 2.5 millions excited electronic structures are calculated based on the time-dependent density functional theory (TD-B3LYP/6-31+G*).
Search PubChemQC
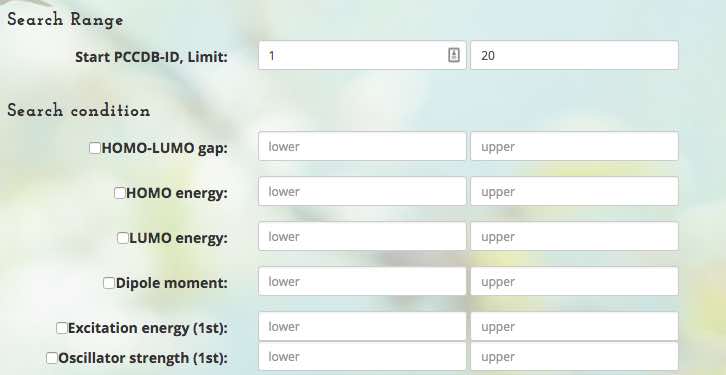
You can search for molecules registered in PubChemQC by using electronic structure properties as keys.
In the ordinary quantum chemistry method, at first a target molecule is specified, and then its electronic structure is calculated. Conversely, in the molecule search approach, we can find several molecules with target properties. In other words, we can solve "inverse problems" by using our systems.
At present, about 2.0 millions molecules composed of H, C, N, O, S, P, and Si atoms can be handled in the system. To provide this search system, we checked calculation data in PubChemQC, because the raw PubChemQC data contains some errors and bugs.
Currently, this search system has some restrictions due to limited server resources. These restrictions will be relaxed in the near future.